Though LAMMPS provides pre-built packages for Windows, the computational efficiency can hardly meet the requirements of large-scale molecular dynamics (MD). A proper strategy to conduct MD on personal computers with Windows systems is to run LAMMPS on Windows Subsystem for Linux (WSL). This article records the process that I compiled LAMMPS with OpenMP and GPU acceleration on WSL2.
Device Infomation
Here’s the information of hardwares and environments of my machine:
- CPU: 13th Gen Intel(R) Core(TM) i7-13650HX (14 cores, 20 threads)
- GPU: NVIDIA GeForce RTX 4060 Laptop GPU
- OS: Ubuntu 22.04 (WSL2 on Windows 11 23H2)
Compiling Process
Preparing Dependencies
build-essential, cmake, and gfortran are needed to compile LAMMPS:
sudo apt install build-essential
sudo apt install cmake
sudo apt install gfortran
sudo apt update
sudo apt upgradeMPICH is neede for parallel computing, and the download link of stable release can be found here.
Use the provided link to download the latese version. Take version 4.2.2 as an example:
wget https://www.mpich.org/static/downloads/4.2.2/mpich-4.2.2.tar.gz ## Version 4.2.2 for exampleInstall MPICH:
tar -xzvf mpich-4.2.2.tar.gz
cd mpich-4.2.2/
./configure
make -j 6 ## Use 6 CPUs for compiling, note that too many CPUs may lead to errors
make installPreparing CUDA Toolkits
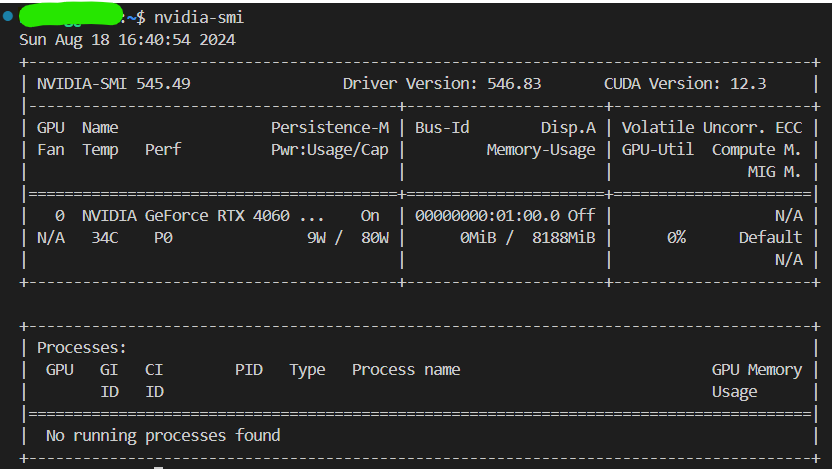
The following command can be used to figure out if there’s any driver for GPU already installed:
nvidia-smiIf a driver has been installed, the information can be seen, and the CUDA version is shown:
Use following command to see if the CUDA toolkits are installed:
nvcc --versionIf the toolkits are installed, the information can be seen:
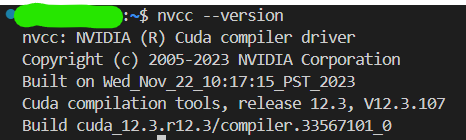
It should be noted that, the version shown here can not be newer than the CUDA version shown in nvidia-smi
If the CUDA has not been installed, follow the instruction from NVIDIA for installation. Take CUDA 12.3 as an example:
wget https://developer.download.nvidia.com/compute/cuda/repos/wsl-ubuntu/x86_64/cuda-keyring_1.1-1_all.deb
sudo dpkg -i cuda-keyring_1.1-1_all.deb
sudo apt-get update
sudo apt-get -y install cuda-toolkit-12-3 ## CUDA Version 12.3Add the CUDA folder (usually at /usr/local/cuda-xx.x/) into PATH, in the ~/.bashrc file:
export PATH=$PATH:/usr/local/cuda-12.3/bin ## For CUDA Version 12.3
export LD_LIBRARY_PATH=$LD_LIBRARY_PATH:/usr/local/cuda-12.3/lib64 ## For CUDA Version 12.3Activate the PATH:
source ~/.bashrcThen you can use the following commands to ensure the installation:
nvidia-smi
nvcc --versionCompiling LAMMPS
Get the latest stable release of LAMMPS:
wget https://download.lammps.org/tars/lammps-stable.tar.gzPrepare for compiling:
tar -xzvf lammps-stable.tar.gzTake the 2Aug2023 version as an example:
cd lammps-2Aug2023/
mkdir build
cd build
In addition to basic packages (MANYBODY, KSPACE, MOLECULE, and RIGID), the OPENMP and GPU packeges can be compiled together:
cmake -C ../cmake/presets/basic.cmake -D PKG_OPENMP=yes -D PKG_GPU=on -D GPU_API=cuda -D GPU_ARCH=sm_89 ../cmake ## code sm_89 for RTX 4060, the coresponding code should be used acoording to the GPU architecture
make -j
# or makeBesides these packages, additional packages can be installed if you need.
The compiled excutable file can be found in this build folder if the process goes properly. This folder can be added into PATH for convenience. In the ~/.bashrc file:
export PATH=$PATH:/path_to_home/lammps-2Aug2023/build
export OMP_NUM_THREADS=2 ## Preset the number of OpenMP threads per MPI taskBench Test
I conducted molecular dynamics emcompassing 1000 water molecules at the NPT ensemble for 10 ps to invistigate the efficiency of OpenMP and GPU acceleration:
units real
dimension 3
boundary p p p
atom_style full
pair_style lj/class2/coul/long 12.000
#pair_modify shift yes
bond_style class2
angle_style class2
dihedral_style class2
improper_style class2
kspace_style ewald 1.0e-4
read_data 1000-H2O.data
neighbor 2.0 bin
neigh_modify every 1 delay 0 check no
velocity all create 300.0 45267
dump 1 all custom 10000 npt_equilibrium.xyz id mol type x y z ix iy iz
fix 1 all npt temp 300 300 100 iso 1.0 1.0 1000
thermo 100
thermo_style custom step temp press density etotal epair ebond eangle edihed pxx pyy pzz lx ly lz
reset_timestep 0
timestep 1
dump trj1 all atom 100 npt_equilibrium.lammpstrj
run 10000
undump 1
unfix 1
undump trj1
write_data npt_equilibrium10ps.data
Notice
package
CLASS2is needed for class 2 force filed. And the 1000-H2O.data file can be found here.
The results:
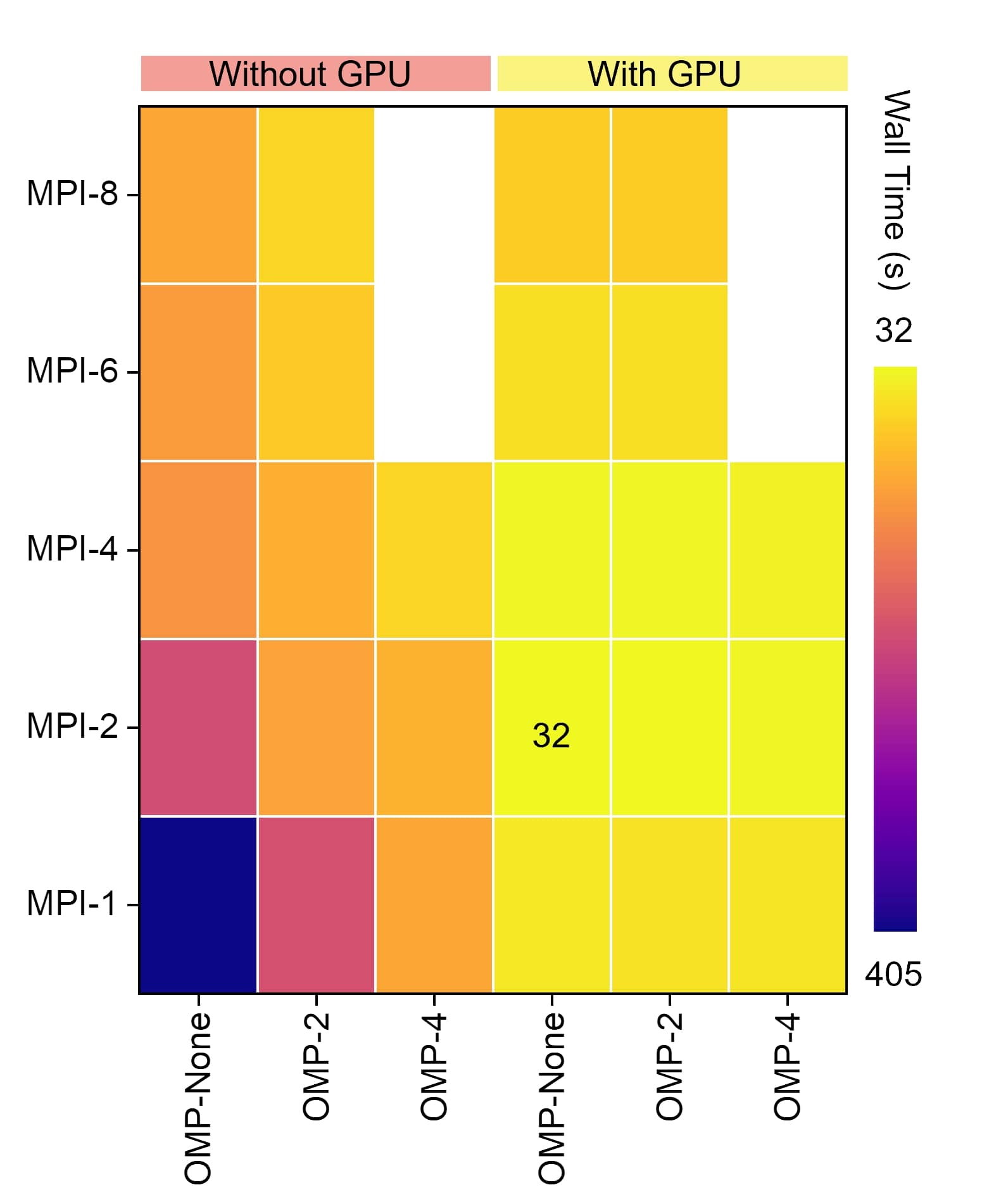 The computation efficiency using OpenMP and GPU is much beter than the ordinary setup. But using GPU with multi-processes and muti-threads may lead to decreased efficiency due to the increased cost of CPU communication.
The computation efficiency using OpenMP and GPU is much beter than the ordinary setup. But using GPU with multi-processes and muti-threads may lead to decreased efficiency due to the increased cost of CPU communication.